RESEARCH PROJECTS
Defining biomarkers of “BRCAness” deficiency in the hereditary breast and ovarian cancer genes

Figure Legend: The current dogma proposes that chemotherapy directly or indirectly generates DSBs that are sensitizing to BRCA mutant cells that have a DNA repair defect. We propose an alternative framework in which “BRCAness” relates to a lagging strand gap vulnerability due to a defect in gap suppression/repair.
Our recent work indicates that DNA replication single stranded DNA gaps (ssDNA) are the true cause of chemotherapy response in BRCA deficient cancer and therefore define BRCAness (Panzarino et al., Cancer Research and Cong et al., Molecular Cell 2021). Rather than an HRD score (homologous recombination deficiency) that relies on tumors being sensitive to chemotherapy based on DNA repair defects, our findings instead indicate responding tumors will be more accurately identified by a gap score. A key question is whether gaps cause protein changes that could signify gaps. To address this question, we performed a non-biased proteomic based approach. We found that as compared to BRCA-proficient cells, BRCA-deficient cells have distinct protein enrichments and reductions in replicating cells. Likewise, we found that BRCA-deficient cells that gain chemoresistance have a distinct set of protein changes that are also critical for gap suppression and chemoresistance. Thus, these enrichments provide an important set of proteins to target and enhance or restore chemosensitivity. Moreover, these proteins show unique nuclear patterns that are being considered for biomarkers of therapy response or BRCAness.
Targeting the replication gap vulnerability in cancer

Figure Legend: Replication gaps as the true vulnerability in BRCA deficient cancer. The molecular basis for the sensitivity of cells with mutations in BRCA proteins to drugs such as PARP inhibitors (PARPi) is thought to stem from the conversion of single-strand DNA breaks to double-strand breaks during DNA replication (current dogma). This concept is supported by the role of the BRCA proteins in fixing DNA double stranded breaks (DSBs) Rather than pausing or stopping at a road block, disparate DNA lesions/roadblocks could similarly trigger re-priming leaving gaps in the wake of replication that we propose is the sensitizing lesion (Alternative Framework). This concept is supported by the role of the BRCA proteins in preventing and fixing single stranded DNA breaks/gaps. If gaps are the true vulnerability, gap making drugs should selectively kill BRCA deficient cancer cells, lets start testing!
As described above, if gaps are the true vulnerability in cancer, then drugs making more gaps should be ideal for uniquely targeting cancer cells. To test this hypothesis, we are addressing whether drugs enhancing lagging strand gaps will sensitize BRCA deficient cancer cells that have an inherent lagging strand synthesis defect. In addition, we are testing the efficacy of targeting gap suppression and repair pathways that operate during or in post-replication. For example, we found that the pathway of translesion synthesis (TLS) is uniquely active in several cancers that have rewired their replisome to engage TLS as a mechanism to suppress gaps and limit cell death. Therefore, TLS is a selective target to kill cancer cells and not harm non-cancer cells (Nayak et al., Science Advances 2020). To systematically address the potential of drug synergy, we are developing a rapid gap detection assay.
Defining why PARP1 activation requires the FANCJ helicase to kick off proteins engaging in mismatch repair
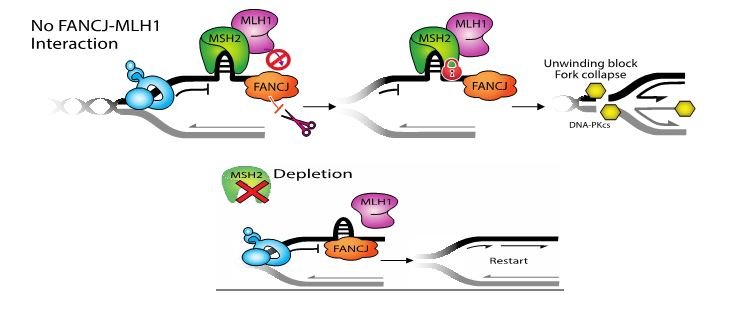
Figure Legend: FANCJ binds directly to the mismatch repair (MMR) protein, MLH1 and this interaction really matters for DNA replication. So far, our data indicate that the interaction helps peel the upstream MSH2-complexes off of DNA and this allows replication to go again. As evidence, we find depletion of MSH2 restores replication restart to cells lacking the FANCJ-MLH1 interaction. Does MSH2 physically blocks replication, or does it block a replication factor? We hope to find out! Could colon cancer due to MMR mutations have replisome changes that provide new opportunities for targeting.
FANCJ null cells display relatively normal DNA replication, but the level of gaps and S-phase PARP1 activity (PARylation) is far below FANCJ proficient cells (Cong et al., Molecular Cell 2021). By re-evaluating the distinct replisome associated changes that develop in FANCJ null cells (Peng et al., Cell Reports 2018), we have developed a hypothesis as to why gaps are absent and why PARP1 is not properly activated. Are findings suggest that gaps and PARP1 activation requires FANCJ to displace mismatch repair (MMR) proteins that interfere with lagging strand synthesis. Now we seek to know how MMR disrupts lagging strand synthesis. This research could provide novel insight into how MMR protects the genome and how to target cancer with loss of MMR as in hereditary colon cancer.
Could gap suppressing drugs help patients with FA?

Figure Legend: Suppressing the gaps to improve FA patient health. If gaps are the fundamental problem in FA, not failed repair of interstrand crosslinks as often proposed, drugs suppressing gaps could be improve FA patient health
Fanconi anemia (FA) is caused by mutations in one of at least 23 FA complementation (FANC) group genes. FA patients display congenital anomalies, hematopoietic stem cell (HSC) injury with increases bone marrow failure and leukemia, and high risk of secondary cancers. The most common feature of cells with FA gene mutations is sensitivity to drugs making DNA interstrand cross links (ICLs) propelling many studies into how FA proteins repair ICLs. However, it is not clear that failed ICL repair underlies FA phenotypes. Indeed, FA cells also have an underlying replication gap problem. Could suppression of gaps alone improve the fitness of FA cells or the health of FA patients? With this hypothesis in mind, we are screening for compounds that suppress gaps. This work is important because mutations underlying FA affect every cell in the body and there is currently no cure for FA.
Hunting down the cancer vulnerability that defines curability

Figure Legend: What is the elusive factor (s) that makes some cancers curable?
Chemotherapy therapy that truly eradicates cancer is restricted to a limited set of malignancies. The underlying feature that defines curability is unknown. Some argue that there is a correlation between chemotherapy curability and genetic events that enable cells to modify their genomes as required during development. This is an intriguing hypothesis because activities that re-arrange the genome also correlate with a high level of apoptosis, a cell death process. Accordingly, cancer derived from cells actively developing could readily undergo apoptosis following chemotherapy. Our goal is to test this hypothesis by addressing if inhibition vs promotion of apoptosis will derail and enhance the success of chemotherapy, respectively. Identifying drug combinations that kill cancer cells but with low toxicity to normal cells is essential for cancer patient survival and quality of life. Can we cure childhood leukemia but also avoid toxic chemotherapy and the threat of future cancer?